SEIR-Modell

Als SEIR-Modell bezeichnet man in der mathematischen Epidemiologie einen Ansatz zur Beschreibung der Ausbreitung von ansteckenden Krankheiten. Die Beschreibung ist näher am realen Verlauf als die des SIR-Modells, da hier berücksichtigt wird, dass ein Individuum nach seiner Infektion nicht sofort selbst infektiös ist. Im Gegensatz zu einem Individuum-basierten Modell ist die Beschreibung makroskopisch, d. h. die Population wird als Gesamtheit betrachtet.
Klassisches SEIR-Modell
Modelldefinition
Die Population von N Individuen sei zerlegt in die vier Kompartimente S, E, I, R, so dass

Jedes Individuum kann die Prozedur
- Susceptible (S) → Exposed (E) → Infectious (I) → Recovered (R)
durchlaufen. Die Ausbreitungsdynamik der Krankheit wird beschrieben durch das System

wobei

die Ableitungen nach der Zeit sind. Betrachtet man die bezüglich der konstanten Population relativen Anteile

nimmt das System die Gestalt

an. Hierbei handelt es sich um ein autonomes nichtlineares System von gewöhnlichen Differentialgleichungen, weshalb das Modell ein dynamisches System beschreibt. Jeder Zustand ohne Exponierte und Infektiöse stellt eine Ruhelage des dynamischen Systems dar, die krankheitsfreies Gleichgewicht genannt wird, zuweilen abgekürzt als DFE für disease free equilibrium. Sollte die effektive Reproduktionszahl den kritischen Wert 1 übersteigen, handelt es sich um eine instabile Ruhelage, aus deren Umfeld sich eine Epidemie entwickelt, woraufhin mit dem Durchlaufen der Epidemie eines der stabilen krankheitsfreien Gleichgewichte angestrebt wird.
| Größe | Einheit | Erklärung |
|---|---|---|
| S(t) | indv | Anteil der Anfälligen, engl. susceptible. Noch nicht infiziert. |
| E(t) | indv | Anteil der Exponierten, engl. exposed. Infiziert, aber noch nicht infektiös. |
| I(t) | indv | Anteil der Infektiösen, engl. infectious. |
| R(t) | indv | Anteil der Erholten, engl. recovered oder resistant. Bzw. verstorben oder nach Symptomen in Quarantäne. |
| t | d | Zeit in Tagen, engl. time. |
| β | 1/d | Transmissionsrate. Der Kehrwert ist die mittlere Zeit zwischen Kontakten mit Übertragung. |
| γ | 1/d | Erholungsrate. Der Kehrwert ist die mittlere infektiöse Zeit. |
| α | 1/d | Übergangsrate. Der Kehrwert ist die mittlere Latenzzeit. |
Die mittlere Latenzzeit ist die durchschnittliche Zeit, die ein Individuum in der Gruppe E der Exponierten verbringt; diese ist zu unterscheiden von der mittleren Inkubationszeit, denn der Beginn der Infektiosität muss nicht mit dem Beginn der Symptome übereinstimmen.
Für die Transmissionsrate (Übertragungsrate) ist auch die Bezeichnung Kontaktrate geläufig. Eine genauere Überlegung zerlegt diese in ein Produkt , wobei die Transmissionswahrscheinlichkeit und die eigentliche Kontaktrate ist.



Modellannahmen
Das Modell macht eine Reihe von Annahmen, welche die Wirklichkeit stark vereinfachen. Im Einzelnen:
- Es findet eine gute Durchmischung der Bevölkerung statt, so dass jedes mit jedem anderen Individuum in Kontakt kommen kann. Andernfalls müsste man Kontaktnetzwerke oder räumliche Dynamiken beschreiben.
- Es gibt hinreichend viele Infektiöse, so dass der Verlauf der Epidemie näherungsweise deterministisch stattfindet. Andernfalls müsste man einen stochastischen Prozess beschreiben.
- Die Werte sind kontinuierlich. Das Modell beschreibt keine diskreten Anzahlen.
- Sowohl die Dauer der latenten Phase als auch der infektiösen Phase ist exponentialverteilt (siehe den Abschnitt Infektionsalter-Modell unten). Die Transmissionsrate in der infektiösen Phase ist hierbei konstant.
- Die Krankheit führt entweder zur vollständigen und lebenslangen Immunität oder zum Versterben.
- Jedes anfällige Individuum ist gleich anfällig für die Krankheit.
- Ein Nachwuchs neuer Anfälliger und natürliches Versterben tritt nicht auf. Das Modell enthält keine Beschreibung der demografischen Dynamik. Insbesondere ist die Größe der Bevölkerung konstant.
Beziehung zur Basisreproduktionszahl
Zwischen den Parametern des Modells und der Basisreproduktionszahl lässt sich eine Beziehung herstellen. Damit sich die Krankheit nicht weiter ausbreitet, muss und sein. Einsetzung dieser Bedingungen in die Differentialgleichungen führt zu und , und somit . Diese Gleichgewichtsbedingungen entsprechen der effektiven Reproduktionszahl . Weil aber per Definition gilt, ergibt sich[1]








Gelegentlich wird alternativ so definiert, dass gilt, mit der Bewandtnis, bei Benutzung der Großbuchstaben den Parameter nicht in den Gleichungen mitführen zu müssen.[2][3] Unterscheidungen dieser Art sind durch Normalisierung auf entfernbar, denn das Modell kodiert per se keine diskreten Zahlen.




Man definiert , das ist der immune Anteil der Population. Im Gleichgewicht


ist demnach

Dieser Anteil, oberhalb dessen Herdenimmunität besteht, heißt kritische Immunisierungsschwelle. Diese Schwelle ist nur von der Basisreproduktionszahl abhängig, nicht aber vom gewählten Modell.
Der Parameter , der den Übergang in das Zwischenstadium beschreibt, in der die Person infiziert, aber noch nicht ansteckend ist, spielt bei der Basisreproduktionszahl keine Rolle, solange man die Möglichkeit des Versterbens in der Gruppe der Exponierten vernachlässigt.[2] Falls man dies nicht tut, erhält man eine Formel für wie unten im Abschnitt Einbeziehung demografischer Dynamik angegeben.


Die Beziehung der Parameter zur Basisreproduktionszahl ist also im Allgemeinen für jede Modifikation des Modells neu zu ermitteln. Eine systematische Methode der Bestimmung wurde im Jahr 1989 von Odo Diekmann, Hans Heesterbeek und Hans Metz gefunden,[4] und später von Pauline van den Driessche und James Watmough weiter ausgearbeitet.[5] Bei dieser Herangehensweise wird die Jacobi-Matrix des Systems am krankheitsfreien Gleichgewicht dergestalt zu einer Differenz zerlegt, dass der Minuend die Neuinfektionen charakterisiert und der Subtrahend die sonstigen Übergänge zwischen den Kompartimenten. Das Produkt aus dem Minuend und der Inverse des Subtrahenden bildet eine neue Matrix, von ihnen Next generation matrix genannt. Ihr Spektralradius ist die Basisreproduktionszahl.
Maximal möglicher Anteil an Infizierten
Die Basisreproduktionszahl bestimmt wesentlich den maximal möglichen Anteil von Infizierten an einer gegebenen Population. Bezeichnen wir den Gesamtanteil aller Infizierten mit , also , so folgt aus dem obigen Differentialgleichungssystem


- ,

oder mit der Basisreproduktionszahl einfach


Mit Umstellung dieser Gleichung nach liefert eine Integration durch Trennung der Variablen


für alle t, wobei der natürliche Logarithmus ist. Insbesondere gilt damit


oder äquivalent

Die Hilfsfunktion hat wegen ein Maximum bei . Der maximal mögliche Anteil der Infizierten wird also für erreicht, d. h. hängt nur von der Basisreproduktionszahl und den Anfangswerten von und ab:







Für eine neu auftretende Erkrankung wie eine Epidemie durch ein unbekanntes Virus gilt und , d. h. der maximal mögliche Anteil der Infizierten an der Population hängt in diesem Fall wie folgt von der Basisreproduktionszahl ab:



Diese Gleichung entspricht der unter SIR-Modell #Maximale Zahl der Infizierten angegebenen Gleichung für das SIR-Modell.
Ausmaß der Epidemie nach ihrem Abklingen
Im Zusammenhang mit der Basisreproduktionszahl steht auch, welcher Anteil der Population insgesamt infiziert wird, unter Annahme, die Epidemie würde ohne jegliche Quarantäne durchlaufen. Unter Heranziehung der Differentialgleichungen findet man

Für den Anfangswert ist demnach


Bei ist nun , und daher . Daraus resultiert die Gleichung




Dieser Zusammenhang, Final size equation genannt, besitzt eine über das SEIR-Modell hinausgehende Bedeutung. Algebraische Umformung führt zur Gleichung

Mit und lautet die letzte Gleichung , die mit der lambertschen W-Funktion nach umgestellt werden kann, d. h. , was wieder zurückersetzt und nach umgestellt








ergibt.
Für die praktische Berechnung des hier relevanten Teils der W-Funktion betrachtet man das quadratische Taylorpolynom der Funktion an der Stelle −1 und bestimmt von diesem die Nullstelle, auf welche noch einmalig die Fixpunktiteration angewendet wird. Das Resultat ist



Eine gute Näherung der W-Funktion erhält man nun als

wobei die n-te Iteration des Newtonverfahrens


ist. Für alle praktischen Bedürfnisse ist völlig ausreichend.

Exponentielle Anfangsphase
Am Anfang der Epidemie verläuft die Ausbreitung der Krankheit in guter Näherung exponentiell. Eine Untersuchung dieser anfänglichen Phase gelingt mit der folgenden systematischen Methode, die auch auf andere Modelle anwendbar ist. Der Vektor fasst die vier Anteile zusammen, so dass das autonome System in der abstrakten Form dargestellt werden kann. An einem Zustand nähert die Linearisierung




das System an, wobei mit die Jacobi-Matrix gemeint ist. Der Anfang der Epidemie liegt am krankheitsfreien Gleichgewicht , das als Ruhelage die Gleichung erfüllt, also eine Nullstelle des Vektorfeldes ist. Die Jacobi-Matrix besitzt in dieser Ruhelage den Wert





Die vierte Zeile des Systems darf entfallen, da sie von den übrigen entkoppelt vorliegt, womit sich die Jacobi-Matrix auf drei Zeilen und Spalten verkleinert. Im linearisierten System ist zudem die erste Zeile entkoppelt, womit eigentlich die Betrachtung des kleinen Systems

genügt. Die allgemeine Lösung des linearisierten Systems erhält man mit Standardmethoden. Man kann nachrechnen, dass die Jacobi-Matrix für immer diagonalisierbar ist. Die Lösung ist demzufolge von der Form , wobei der Eigenvektor zum Eigenwert ist und die Konstanten aus der Anfangsbedingung zu ermitteln sind.





Diese allgemeine Lösung gilt auch für sonderbare Anfangswerte. Einer der Summanden fällt jedoch rasch auf null ab, weshalb man ein wenig später für die Neuinfektionen eigentlich nur eine exponentielle Kurve beobachtet, womit sich die Lösungen ebenfalls zu Exponentialfunktionen vereinfachen. Das Verhältnis von Exponierten zu Infizierten strebt mit dem Abfallen des Summanden einen speziellen Wert an. Weil infolge aufgrund gilt, finden sich die natürlichen Anfangswerte




Weiterhin ergibt sich nun, dass die Wachstumskonstante als Eigenwert der Jacobi-Matrix die quadratische Gleichung


erfüllen muss, womit eine Beziehung zwischen der Wachstumskonstante und den Parametern des Modells gefunden ist.[6] Wie bei jedem exponentiellen Wachstumsvorgang steht die Wachstumskonstante in eins-zu-eins-Beziehung zur anschaulicheren Vervielfachungszeit

speziell zur Verdopplungszeit .

Die Linearisierung an der Ruhelage ist überdies für die Untersuchung der Stabilität von Bedeutung. Besitzt die Jacobi-Matrix keinen Eigenwert mit Realteil null, spricht man von einer hyperbolischen Ruhelage. Laut dem Linearisierungssatz, einem Grundresultat der Stabilitätstheorie dynamischer Systeme, besitzt das linearisierte System in der Nähe der hyperbolischen Ruhelage dasselbe qualitative Verhalten wie das ursprüngliche. Somit ist eine hyperbolische Ruhelage genau dann stabil, wenn sich sämtliche Eigenwerte links der imaginären Achse befinden. Weil die vorliegende Jacobi-Matrix allerdings den Eigenwert null enthält, müsste man weitergehende Mittel zur Untersuchung der Stabilität einsetzen. Eine Umgehung dieses Problems bietet das später beschriebene Modell mit demografischer Dynamik im speziellen Fall, wo die Geburtenrate mit der Sterberate übereinstimmt. Dann ist N konstant bezüglich der Zeit, womit man das äquivalente System definieren darf, in dem N eine explizite Konstante ist. Der Eigenwert null tritt nun nicht mehr auf. Damit einhergehend verbleibt aber aufgrund des Versterbens der Resistenten nur noch die vollständig anfällige Population als einziges krankheitsfreies Gleichgewicht. Ferner tritt im instabilen Fall das stabile endemische Gleichgewicht in Erscheinung, das im einfachen Modell nicht existiert.
Die Untersuchung zur Stabilität wurde von van den Driessche in allgemeiner Weise durchgeführt. Sie kam zum Resultat, dass das krankheitsfreie Gleichgewicht bei einem deterministischen Kompartiment-Modell unter gewissen natürlichen Voraussetzungen an die Modelldefinition erwartungsgemäß immer dann instabil ist, wenn die Basisreproduktionszahl über dem kritischen Wert 1 liegt, und zumindest lokal asymptotisch stabil, wenn sie darunter liegt. Ebenso findet man bei Erläuterungen dieser Untersuchung aufgrund der besagten Problematik zunächst die Beschränkung auf solche Modelle vor, wo ein einziges krankheitsfreies Gleichgewicht existiert.
Beispielrechnung
Es folgt eine Beispielrechnung zu einer Parameterbelegung, wie sie im März 2020 für die COVID-19-Pandemie in Deutschland abgeschätzt wurde.[7] Die Ausbreitung verläuft bezüglich einer Basisreproduktionszahl von 2,4, was der Unterlassung wesentlicher Quarantäne entspricht. Zur numerischen Lösung des Anfangswertproblems genügt das Euler-Verfahren.
from numpy import array as vector # Explizites Euler-Verfahren def euler_method(f, t0, x0, t1, h): t = t0; x = x0 a = [[t, x]] for k in range(0, 1 + int((t1 - t0)/h)): t = t0 + k*h x = x + h*f(t, x) a.append([t, x]) return a def SEIR_model(alpha, beta, gamma): def f(t, x): s, e, i, r = x return vector([ -beta*s*i, beta*s*i - alpha*e, alpha*e - gamma*i, gamma*i ]) return f def SEIR_simulation(alpha, beta, gamma, e0, i0, days, step=0.1): x0 = vector([1.0 - e0 - i0, e0, i0, 0.0]) f = SEIR_model(alpha, beta, gamma) return euler_method(f, 0, x0, days, step) def diagram(simulation): import matplotlib.pyplot as plot plot.style.use('fivethirtyeight') figure,axes = plot.subplots() figure.subplots_adjust(bottom = 0.15) axes.grid(linestyle = ':', linewidth = 2.0, color = "#808080") t,x = zip(*simulation()) s, e, i, r = zip(*x) axes.plot(t, s, color = "#0000cc") axes.plot(t, e, color = "#ffb000", linestyle = '--') axes.plot(t, i, color = "#a00060") axes.plot(t, r, color = "#008000", linestyle = '--') plot.show() def simulation1(): N = 83200000 # Einwohnerzahl von Deutschland 2019/2020 R0 = 2.4; gamma = 1/3.0 return SEIR_simulation( alpha = 1/5.5, beta = R0*gamma, gamma = gamma, e0 = 40000.0/N, i0 = 10000.0/N, days = 140) diagram(simulation1)
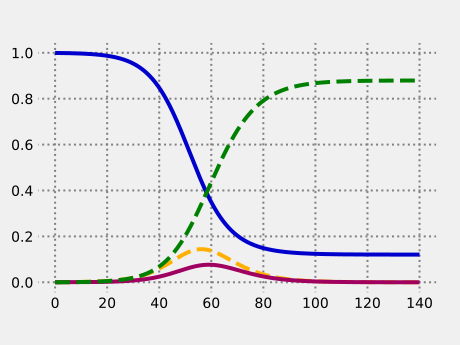 Die vier Anteile, jeweils abhängig von der Zeit in Tagen. |
Ersichtlich ist an diesem Beispiel, dass die Epidemie aufgrund der Aufheizung durch die Infektiösen auch noch nach dem Erreichen der kritischen Immunisierungsschwelle

weiterläuft. Insgesamt würden sich 88 % der Bevölkerung mit der Krankheit anstecken. Die Epidemie ließe sich allerdings spätestens nach Erreichen der kritischen Immunisierungsschwelle durch eine ca. einmonatige strenge Quarantäne stoppen.
Modellerweiterungen
Einbeziehung demografischer Dynamik

Unter der Annahme einer konstanten Geburtenrate und konstanten Sterberate wurde das erweiterte Modell



aufgestellt. Gegenüber dem einfachen SEIR-Modell beschreibt dieses Modell auch einen langfristigen endemischen Verlauf, bei dem es zu einer Schwingung des Anteils der Anfälligen kommen kann, bis sich dieser ins endemische Gleichgewicht eingependelt hat. Für verschwindende Latenzzeit geht das Modell ins korrespondierende SIR-Modell über, wo dieses Phänomen bereits auftritt.
Sofern sich die Geburten- und Sterberate voneinander unterscheiden, ist die Populationsgröße

keine Konstante mehr, sondern verläuft gemäß

Die Beziehung zur Basisreproduktionszahl ist

Ist das Wachstum der Bevölkerung oder ihr Schwund über den betrachteten Zeitraum verschwindend gering, kann man die Geburten- und Sterberate zur Vereinfachung gleich setzen. In diesem Fall pendelt sich der Anteil der Anfälligen genau in den Gegenwert der kritischen Immunisierungsschwelle ein.
Zeitabhängige Transmissionsrate
Vorgänge wie Verhaltensänderungen, Quarantäne und Saisonalität bewirken eine Veränderung der Transmissionsrate. Diese Umstände finden ihre Berücksichtigung in der Modellierung der Transmissionsrate als zeitabhängige Funktion , wobei das übrige Modell identisch beibehalten wird.[8] Die einfachsten Ansätze für die Saisonalität nehmen die Transmissionsrate zum Beispiel als Sinusschwingung an, mit Berg in den kälteren und Tal in den wärmeren Monaten.[9]

Zu bemerken ist, dass das Differentialgleichungssystem mit der expliziten Zeitabhängigkeit kein autonomes mehr ist und damit nicht mehr direkt ein dynamisches System vorliegt. Man kann aus dem System allerdings durch Hinzunahme der Gleichung künstlich ein autonomes gewinnen.

Interagierende Subpopulationen
Da soziale Kontakte, Schwere und Letalität der Erkrankung sowie die Wirksamkeit prophylaktischer Maßnahmen sich zwischen interagierenden Subpopulationen deutlich unterscheiden können, werden im „Interacting Subpopulation SEIR model“ individuelle SEIR Modelle für jede Subgruppe konstruiert und durch Kontakt-Interaktionen zwischen den Gruppen verknüpft[10]. Solche Modelle werden zum Beispiel zur Modellierung der Covid-19-Pandemie verwendet, um personalisierte, beschleunigte Impfstrategien zu entwickeln[11], welche eine Verkürzung der Pandemie und eine Reduktion von Fallzahlen und Todesfällen voraussagen.
Verallgemeinerungen
Infektionsalter-Modell
Die im SEIR-Modell vorausgesetzte Exponentialverteilung der Latenzzeit und der infektiösen Zeit mag recht künstlich erscheinen. Ein allgemeines, von dieser Voraussetzung befreites Modell beschreibt den zeitlichen Verlauf des Anteils der Anfälligen durch die Gleichung[12]

Hierbei ist die Infektionsrate im Infektionsalter . Mit dem Infektionsalter ist nicht das Alter eines Individuums gemeint, sondern die seit der Infektion vergangene Zeit. ist die Zeitableitung von .




Das Modell ist wie folgt interpretierbar. Ursache für die Neuinzidenz sind in der Vergangenheit infizierte Infektiöse. Um in der Zeit zurück kam hinzu, die sich nun im Infektionsalter befinden und daher zu beitragen. Das Integral über die gesamte Vergangenheit ist schließlich noch mit zu multiplizieren, weil zum aktuellen Zeitpunkt nur noch der Anteil anfällig ist.



Per Definition von ergibt sich die Basisreproduktionszahl nun gemäß der Formel

Die normierte Funktion lässt sich als Dichte einer Wahrscheinlichkeitsverteilung auffassen, die man Verteilung der Generationszeit, englisch generation interval distribution, nennt. Ihr Erwartungswert, die Generationszeit, ist der mittlere Abstand zwischen dem Zeitpunkt der Infektion des Spenders und dem Zeitpunkt der Infektion des Empfängers der Krankheit. Im Modell bleibt die Final size equation gültig, womit das Ausmaß der Epidemie allein von der Basisreproduktionszahl abhängt, nicht jedoch von der Dichte.

Zur Anfangsphase der Epidemie ergibt sich aus dem Modell für eine vollständig anfällige Population ( in guter Näherung) mit dem Ansatz zudem die epidemiologische Euler-Lotka-Gleichung



Diese besagt, bei Kenntnis der Dichte und der Wachstumskonstante ist die Basisreproduktionszahl ohne Kenntnis der Infektionsrate ermittelbar.
Um zu verstehen, in welchem Bezug die Modelle stehen, zerlegt man die Infektionsrate in das Produkt , wobei die Transmissionsrate ist, und die Wahrscheinlichkeit infektiös zu sein. Im Folgenden ist die Transmissionsrate konstant. Beginnt die infektiöse Zeit sofort und ist deren Dauer zufällig verteilt gemäß der Verteilungsfunktion , gilt . Speziell für ergibt sich das SIR-Modell, wobei die mittlere infektiöse Dauer nichts anderes als der Erwartungswert der Verteilung ist.







Dauert nun die Latenzzeit zufällig lang gemäß Verteilungsfunktion , und die danach beginnende infektiöse Zeit zufällig lang gemäß Verteilungsfunktion , erhält man




Speziell für und ergibt sich das SEIR-Modell. Die Erwartungswerte sind die mittlere Latenzzeit und die mittlere infektiöse Zeit.


Für einen nicht-zufälligen infektiösen Zeitraum mit Beginn im Infektionsalter und Ende in , also , vereinfacht sich das Modell stattdessen zu


![{\displaystyle p(a)=\chi _{[a_{1},a_{2}]}(a)}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b36a5173c98787d603786484ee9349a4b5003da9)

eine retardierte Differentialgleichung. Die Beziehung zur Basisreproduktionszahl ist dann .

Stochastische Varianten
CTMC-Modell

Zur Beschreibung von Epidemie-Verläufen im Bereich weniger Infizierter wurde zu den klassischen Modellen jeweils eine direkte Entsprechung als zeitkontinuierlicher Markow-Prozess aufgestellt (CTMC: Continuous time Markov chain). In diesen stochastischen Modellen kann eine Epidemie vorläufig zum Erliegen kommen, obwohl die Reproduktionszahl größer als eins ist.
Für einen Markow-Prozess sei wie üblich


die Übergangswahrscheinlichkeit vom Zustand in den Zustand . Für das SEIR-Modell ist und[13]


Ein Verfahren zur Simulation dieser Markow-Prozesse ist der Gillespie-Algorithmus. Gemäß den Gesetzmäßigkeiten von Markow-Prozessen ist die Verweildauer im Zustand exponentialverteilt mit Verteilungsfunktion , wobei die Ereignisrate die Summe der Einzelraten ist, das heißt


Mittels der Inversionsmethode ist für eine Standardzufallszahl die Verweildauer bzw. der Zeitschritt demnach
![{\displaystyle u\in [0,1]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5ab4b2bf4a8cda1b335fdb4f789c61285c971034)

Eine zweite Standardzufallszahl wählt aus, welches der paarweise disjunkten Ereignisse stattfinden soll:
- Eine Infektion geschieht mit Wahrscheinlichkeit ,
- ein Übergang ins Infektiöse mit Wahrscheinlichkeit ,
- eine Erholung mit der restlichen Wahrscheinlichkeit .



DTMC-Modell

Für einen kleinen Zeitschritt entsteht aus dem CTMC-Modell eine Näherung in Form eines zeitdiskreten Markow-Prozesses (DTMC: Discrete time Markov chain). Ist der Zeitschritt hinreichend klein, so dass mit ihm höchstens ein Ereignis stattfindet, darf man den Term ignorieren und erhält die Übergangswahrscheinlichkeiten analog zum CTMC-Modell.[14]


Die Simulation gestaltet sich besonders einfach. Eine Standardzufallszahl wählt aus, welches der paarweise disjunkten Ereignisse im Zeitschritt stattfinden soll:
- Eine Infektion geschieht mit Wahrscheinlichkeit ,
- ein Übergang ins Infektiöse mit Wahrscheinlichkeit ,
- eine Erholung mit Wahrscheinlichkeit ,
- Verharren im Zustand mit der restlichen Wahrscheinlichkeit.



Weblinks
- Modellrechner für den Verlauf einer Epidemie nach dem SEIR-Modell mit änderbaren Input-Variablen (englisch)
Siehe auch
- Mathematische Modellierung der Epidemiologie
- SI-Modell (Ansteckung ohne Gesundung)
- SIR-Modell (Ansteckung ohne Latenzzeit)
- SIS-Modell (Ausbreitung von ansteckenden Krankheiten ohne Immunitätsbildung)
Literatur
- Matt J. Keeling, Pejman Rohani: Modeling Infectious Diseases in Humans and Animals. Princeton University Press, 2008.
- Matthias an der Heiden, Udo Buchholz: Modellierung von Beispielszenarien der SARS-CoV-2-Epidemie 2020 in Deutschland. Robert Koch-Institut, Abteilung für Infektionsepidemiologie (20. März 2020). doi:10.25646/6571.2.
Einzelnachweise
- ↑ Institute for Disease Modeling: Originals vom 7. Mai 2020 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis.@1@2Vorlage:Webachiv/IABot/www.idmod.org (abgerufen am 12. April 2020)
- ↑ a b Pauline van den Driessche: Reproduction numbers of infectious disease models. In: Infectious Disease Modelling, Band 2, KeAi Publishing, August 2017, S. 288–303. doi:10.1016/j.idm.2017.06.002. PMC 6002118 (freier Volltext).
- ↑ Odo Diekmann, Hans Heesterbeek, Tom Britton: Mathematical tools for understanding infectious disease dynamics. Princeton University Press, 2013, S. 35.
- ↑ Odo Diekmann, J. A. P. Heesterbeek, J. A. J. Metz: On the definition and the computation of the basic reproduction ratio R0 in models for infectious diseases in heterogeneous populations. In: Journal of Mathematical Biology, Band 28, 1990, S. 365–382. doi:10.1007/BF00178324.
- ↑ Pauline van den Driessche, James Watmough: Reproduction numbers and sub-threshold endemic equilibria for compartmental models of disease transmission. In: Mathematical Biosciences, Band 180, Nr. 1–2, November–December 2002, S. 29–48. doi:10.1016/s0025-5564(02)00108-6.
- ↑ Junling Ma: Estimating epidemic exponential growth rate and basic reproduction number. In: Infectious Disease Modelling, Band 5, 2020, S. 129–141. doi:10.1016/j.idm.2019.12.009.
- ↑ Stellungnahme der Deutschen Gesellschaft für Epidemiologie (DGEpi) zur Verbreitung des neuen Coronavirus (SARS-CoV-2). (PDF) Deutsche Gesellschaft für Epidemiologie, 18. März 2020, abgerufen am 26. März 2020. Die der Beispielrechnung zugrunde liegende Version vom 19. März ist nicht mehr auffindbar. Hier ersatzweise die Version vom Vortag. Die aktuelle Version siehe hier (Memento des Originals vom 29. Mai 2020 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis.@1@2Vorlage:Webachiv/IABot/www.dgepi.de.
- ↑ Gerardo Chowell, Cécile Viboud, Lone Simonsen, Seyed M. Moghadas: Characterizing the reproduction number of epidemics with early subexponential growth dynamics. In: Journal of the Royal Society Interface, Band 13, Nr. 123, 17. August 2016. doi:10.1098/rsif.2016.0659.
- ↑ M. Keeling, P. Rohani: Modeling Infectious Diseases in Humans and Animals. Abschnitt 5.2. (S. 159): Modeling forcing in childhood infectious diseases: Measles.
- ↑ Patrick Hunziker: Personalized-dose Covid-19 vaccination in a wave of virus Variants of Concern: Trading individual efficacy for societal benefit. In: Precision Nanomedicine. 24. Juli 2021, ISSN 2639-9431, doi:10.33218/001c.26101 (precisionnanomedicine.com [abgerufen am 18. August 2021]).
- ↑ Patrick Hunziker: Vaccination strategies for minimizing loss of life in Covid-19 in a Europe lacking vaccines. Infectious Diseases (except HIV/AIDS), 1. Februar 2021, doi:10.1101/2021.01.29.21250747 (medrxiv.org [abgerufen am 18. August 2021]).
- ↑ Fred Brauer: Mathematical epidemiology: Past, present, and future. In: Infectious Disease Modelling, Band 2, Nr. 2, 2017, S. 113–127. doi:10.1016/j.idm.2017.02.001. PMC 6001967 (freier Volltext).
- ↑ William Tritch, Linda J. S. Allen: Duration of a minor epidemic. In: Infectious Disease Modelling, Band 3, 2018, S. 60–73. doi:10.1016/j.idm.2018.03.002. PMC 6326226 (freier Volltext).
- ↑ Linda J. S. Allen: An Introduction to Stochastic Epidemic Models. In: Fred Brauer, Pauline van den Driessche, Jianhong Wu: Mathematical Epidemiology. In: Lecture Notes in Mathematics, Band 1945. Springer, Berlin/Heidelberg 2008, S. 81–130. doi:10.1007/978-3-540-78911-6 3.









